Research interests
Misregulation of the epigenome is a fundamental mechanism in cancer, which results in corruption of the highly specific transcriptional programs, leading to abnormal cell proliferation and expansion. Recent advances in genome analysis reveal that genetic aberrations in human cancer are extremely complex. While most driver mutations remain undruggable, the dependency on resulting aberrant chromatin states holds great promise for the development of targeted therapies.
The causal relationships between histone and DNA modifications, their influence on transcription and functional DNA element integrity are complex and incompletely understood. A primary challenge in the study of aberrant gene regulation in cancer has been to elucidate how chromatin networks influence the millions of putative regulatory elements throughout the genome, which orchestrate the control of tens of thousands of genes in a highly coordinated and context-specific manner.
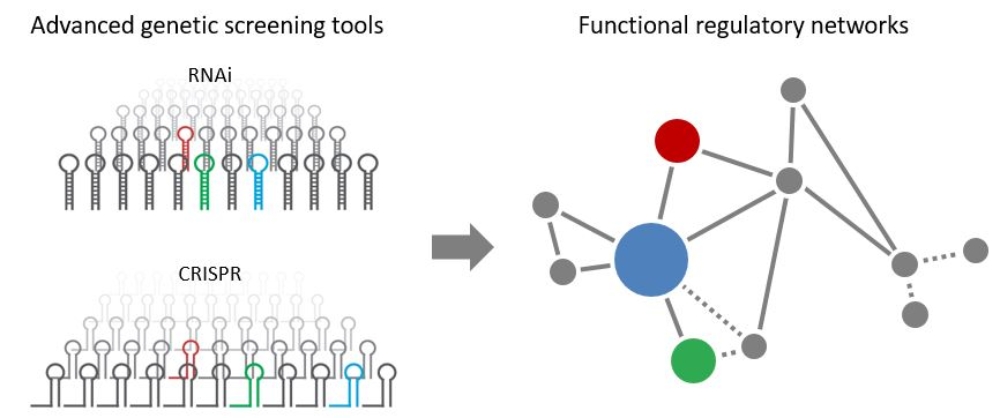
To gain a better understanding of these regulatory epigenetic networks, and ultimately to identify innovative therapeutic strategies and novel biomarkers, we apply a variety of genetic tools, ranging from advanced RNAi to CRISPR/Cas9 technologies combined with tractable fluorescent reporter systems, which are stably integrated into the genome of mouse and human cancer cell lines.
Research lines
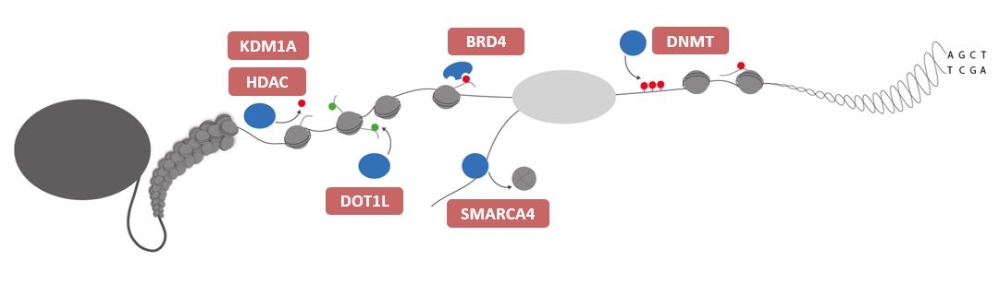
The transcriptional output of effector proteins (e.g. epigenetic effectors, transcription factors or nuclear receptors) is determined by a highly complex network of coregulators. Our limited mechanistic understanding of most of these coregulators complicate the understanding of their function in different cancer contexts. Concurrently, the dependence of effector proteins on accessory proteins gives various opportunities for therapeutic intervention. Traditional biochemical approaches like immunoprecipitation (IP) and chromatin IP (ChIP) have mostly provided a static picture of regulatory chromatin complexes. These approaches have been insufficient to generate a comprehensive understanding of regulatory chromatin networks controlling the expression of essential gene regulatory circuits. We combine fluorescent reporter systems with multiplexed RNAi and CRISPR screening approaches to functionally characterize these diverse complexes. Our initial efforts focus on selected key drug targets and cancer types.
Associated students:
Check out our recent proof-of-principle study published in NAR:
https://academic.oup.com/nar/advance-article/doi/10.1093/nar/gkab180/6212757
Featured summary by the University Stuttgart and the epigentics blog epigenie:
https://www.uni-stuttgart.de/en/university/news/all/publish-genom-research/
https://epigenie.com/removing-r-loops-regulates-repression-revealing-the-role-of-lsd1/
Newsflash from the BioPro BW network:
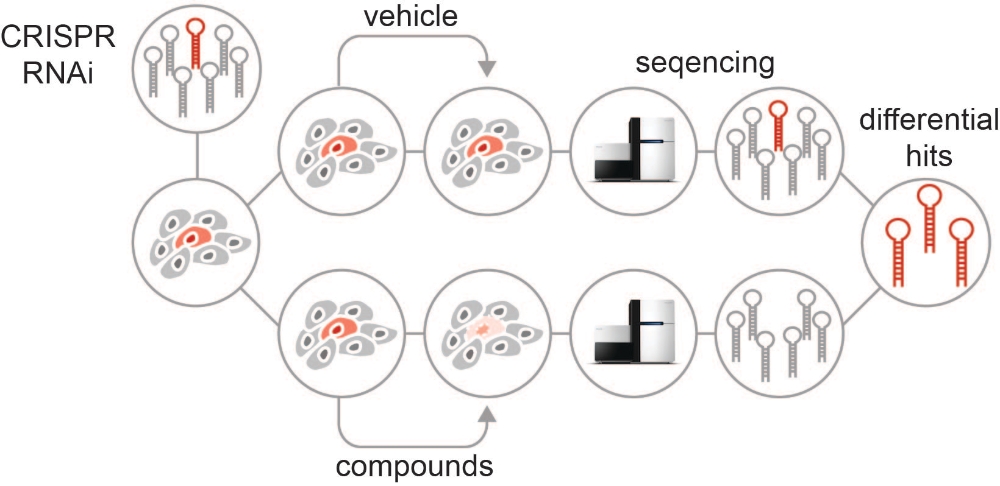
Cancer-specific dependencies on functional DNA elements cannot be predicted from conventional epigenome analyses, but can only be revealed through hypothesis-driven or systematic functional genetic studies. Using multiplexed RNAi screening technologies combined with extensive functional characterization, we have shown in a recent publication (Rathert et. al., 2015) that epigenetic events are crucial for acquired and primary resistance to BET inhibitors (BETis) in leukemia. We are currently expanding this view usingfunctional genetic approaches (CRISPR and RNAi) to other drugs and cancer contexts.
Associated students:
Visit https://www.mdpi.com/2072-6694/13/15/3801 for more information on our recent findings about epigentic resistance mechanisms to chemotherapy in ovarian cancer.

Long-duration orbital space-flights are associated with increased levels of psychological stress, acute and chronic exposure to space radiation and microgravity-induced changes, all of which are known to detrimentally impact the immune system. We hypothesize that stress-induced dysregulation of the human immune system in space may reflect changes in immune cells during malignancies. Findings in the field could lead to alternative cancer therapies.
The immune system is responsible for sensing pathogens and initiating immune responses against infections. Besides that, it is involved in the bidirectional interaction between the gut microbiota and the brain and impacts tumor fate in different stages of disease. Immune cells have complex signaling pathways which are tightly regulated at different levels by epigenetic factors.
Epigenetic changes play fundamental roles in biological and pathological processes by interpreting environmental signals and regulating gene expression. These dynamic expression patterns, from initiation to resolution of the immune response, are due to distinct chromatin states at regulatory DNA sequences e.g. gene promoters and enhancers and it is largely unknown how long-term space-flight influences changes in the epigenome.
The Project is performed in collaboration with:
European Space Agency (ESA)
Funding is provided by the German Aerospace Center (DLR).
Funding

Philipp Rathert
PD Dr.Akademischer Rat und Gruppenleiter