Highlights

Flanking sequence preferences of TET enzymes
Adam et al. (2022) Flanking sequences influence the activity of TET1 and TET2 methylcytosine dioxygenases and affect genomic 5hmC patterns. Communications Biology, 5:92.
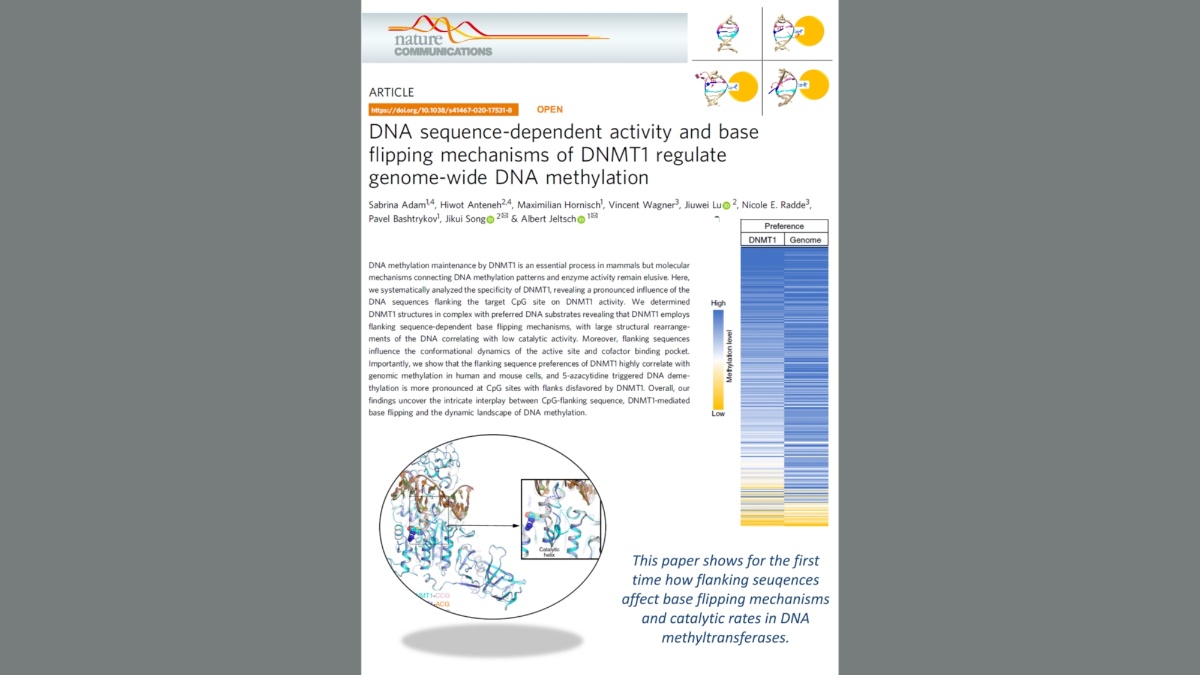
Structure of DNMT1 explains base flipping mechanisms
Adam et al. (2020) DNA sequence-dependent activity and base flipping mechanisms of DNMT1 regulate genome-wide DNA methylation. Nat Commun. 11:3723.

Specificity of SETD2
Schuhmacher et al. (2020) Sequence specificity analysis of the SETD2 protein lysine methyltransferase and discovery of a SETD2 super-substrate. Communications Biology 3, 511.

Structure of DNMT3B explains ICF methylation defects.
Gao et al. (2020) Comprehensive Structure-Function Characterization of DNMT3B and DNMT3A Reveals Distinctive De Novo DNA Methylation Mechanisms. Nat Commun. 11:3355.

Engineering of Effector Domains for Targeted DNA Methylation with Reduced Off-Target Effects
Hofacker, Broche, Laistner, Adam, Bashtrykov & Jeltsch (2020) Int J Mol Sci. 21(2):E502
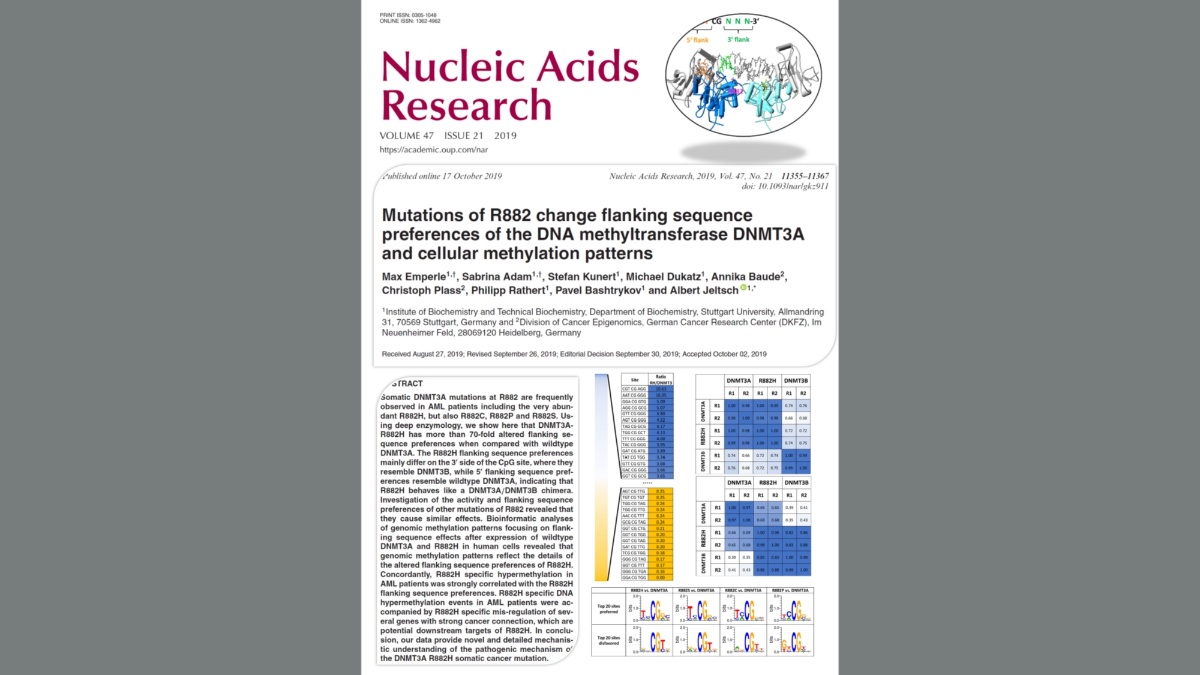
Mutations of R882 change flanking sequence preferences of the DNA methyltransferase DNMT3A and cellular methylation patterns
Emperle, Adam, Kunert, Dukatz, Baude, Plass, Rathert, Bashtrykov & Jeltsch (2019) Nucleic Acids Res. 47(21):11355-11367

The SETDB1 triple Tudor domain binds H3 K9me/K14ac
Jurkowska, Qin, Kungulovski, Tempel, Liu, Bashtrykov, Stiefelmaier, Jurkowski, Kudithipudi, Weirich, Tamas, Wu, Dombrovski, Loppnau, Reinhardt, Min & Jeltsch (2017) Nat Commun. 8(1):2057

Lungu, Pinter, Broche, Rathert & Jeltsch (2017) Nature Communications 8:649
Modular fluorescence complementation sensors for live cell detection of epigenetic signals at endogenous genomic sites.
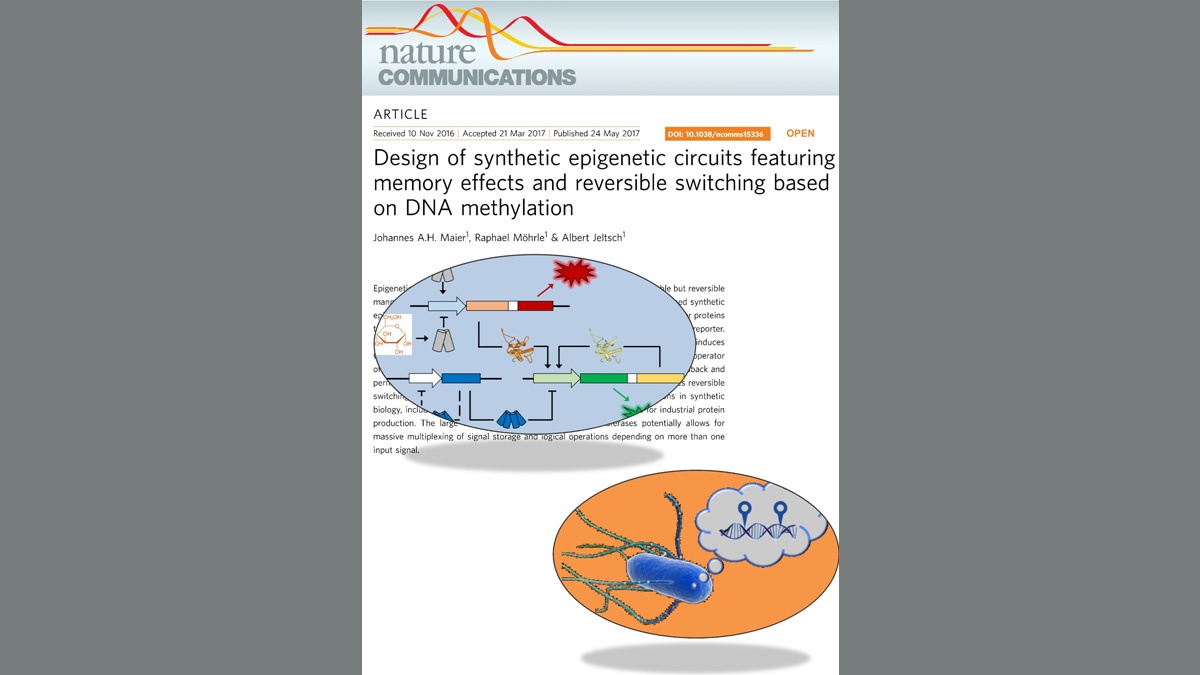
Maier, Möhrle & Jeltsch (2017) Nature Communcations 8, 15336
Design of synthetic epigenetic circuits featuring memory effects and reversible switching based on DNA methylation.

Kudithipudi & Jeltsch (2016) Cell Chem. Biol. 23, 1049-55
Approaches and Guidelines for the Identification of Novel Substrates of Protein Lysine Methyltransferases.

Kungulovski & Jeltsch (2016) Trends in Genetics 32, 101-113
Epigenome Editing: State of the Art, Concepts, and Perspectives.

Kungulovski et al. (2014) Genome Research 24, 1842-53
Application of histone modification-specific interaction domains as an alternative to antibodies. Genome Research 24(11): 1842-53.
Since 2015
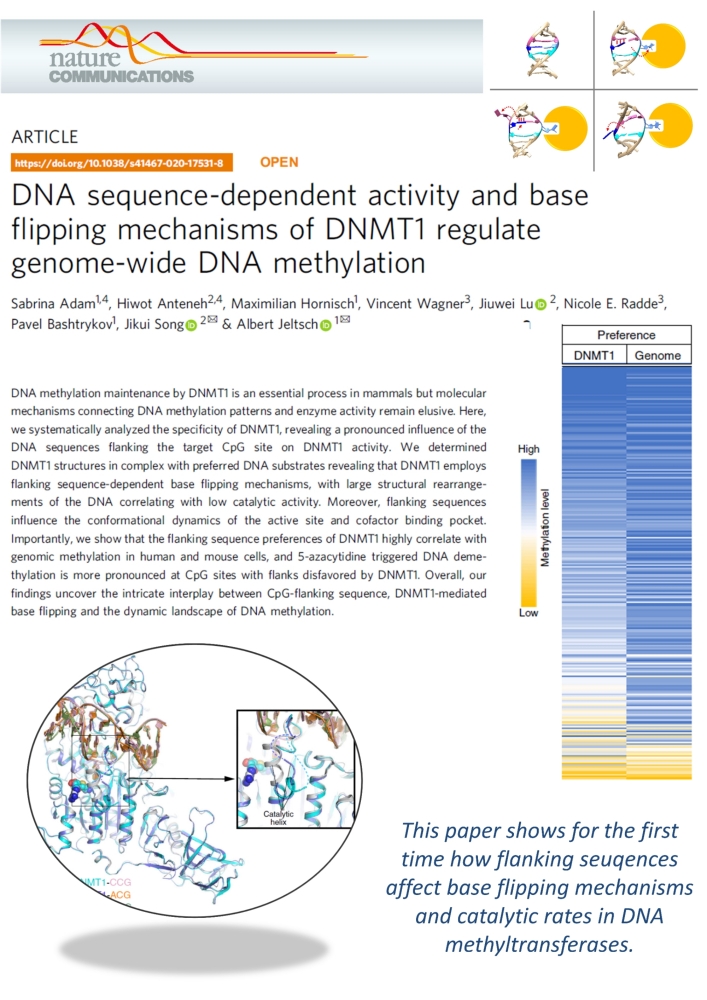
Adam et al. (2020) DNA sequence-dependent activity and base flipping mechanisms of DNMT1 regulate genome-wide DNA methylation. Nat Commun. 11:3723
DNA methylation on CG sequences is a chemical modification of DNA that is essential for human embryonic development. The DNA methyltransferase DNMT1 plays a central role in this process. The enzyme uses a spectacular base flipping mechanism, the basics of which are not well understood. An international research team led by Prof. Albert Jeltsch from the Institute for Biochemistry and Technical Biochemistry (IBTB) at the University of Stuttgart used biochemical and structural biological experiments to clarify the dynamic processes involved in this process.
In 1993 it was shown for the first time that DNA methyltransferases partially destroy the DNA double helix structure discovered by Watson and Crick in 1953 in the course of their catalyzed reaction, because the cytosine base to be methylated is flipped out of the DNA helix. The guanine, originally forming a base pair with the cytosine, is left behind in the DNA helix, which leads to dynamic structural changes in the DNA, which, however, have not yet been systematically investigated.
A team led by Prof. Albert Jeltsch from the University of Stuttgart was able to measure the DNA methylation rate of DNMT1 methyltransferase on thousands of DNA sequences containing CG target sequences using a new method. It was shown that the sequence environment has a significant influence on the enzyme activity. In cooperation with a group from the University of California, structures of DNMT1 with different DNA sequences could be solved, which show that the structural changes of the DNA after base flipping depend directly on the neighboring DNA sequence, and these effects also control the turnover rate of DNMT1 . Complexes with minor structural changes showed a high turnover rate, while a complex with a massive structure change showed a slow turnover rate.
With the help of modeling experiments and simulations in cooperation with the group of Prof. Radde from the University of Stuttgart, the mechanism of the reaction could be described in more detail. Further experiments showed that the DNA methylation pattern in human cells and also the effect of DNMT inhibitors used in cancer treatment are strongly influenced by the sequence dependence of DNMT1 activity. "It is fascinating to see how dynamic processes in biomolecules at the atomic level are reflected in global properties such as genome-wide DNA methylation patterns in human cells and the effects of drugs," says Jeltsch.
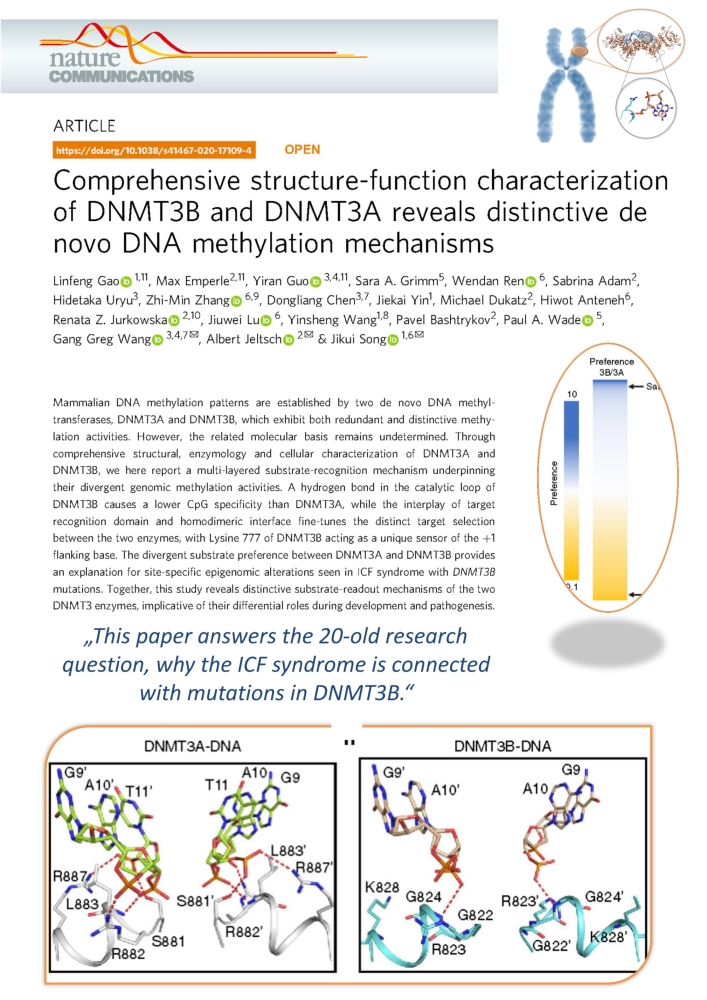
Gao et al. (2020) Comprehensive Structure-Function Characterization of DNMT3B and DNMT3A Reveals Distinctive De Novo DNA Methylation Mechanisms. Nat Commun. 11:3355
In the course of the evolution of higher organisms such as mammals, gene duplication often occurs. The resulting "twin genes" have a very similar genetic makeup, but are independent of each other and can therefore specialize in certain tasks. An example of this is DNA methylation, a chemical change in the basic building blocks of the genetic material of a cell, which is caused by the transfer of methyl groups by enzymes (DNA methyl transferases, DNMT) to certain locations in the DNA. The DNA-methyltransferases DNMT3A and DNMT3B are two forms of these enzymes in human cells that have arisen from gene duplication.
Over 20 years ago, it was discovered that DNMT3B is essential for the DNA methylation of certain regions in the human genome and that insufficient activity of DNMT3B leads to the so-called ICF syndrome. However, why DNMT3B is specifically required for this task and why, for example, the twin DNMT3A cannot take over this task has so far remained unknown.
DNMT3B is required for the methylation of certain sequences in human chromosomes (orange). The specificity of DNMT3B for these regions is determined by a specific protein loop around Arginine 823 (gray). Figure: University of Stuttgart / IBTB
In cooperation with groups from the University of California and the University of North Carolina at Chapel Hill, our team has now been able to solve the structure of DNMT3B in complex with other DNA sequences and measure the turnover rate of DNA methylation by DNMT3B and DNMT3A on thousands of DNA sequences. It was shown that DNMT3B is particularly active on its target sequences in the human genome due to a special protein loop, while DNMT3A can only work poorly on these sequences.

Hofacker, Broche, Laistner, Adam, Bashtrykov & Jeltsch (2020) Engineering of Effector Domains for Targeted DNA Methylation with Reduced Off-Target Effects. Int J Mol Sci. 21(2)
Epigenome editing is a promising technology, potentially allowing the stable reprogramming of gene expression profiles without alteration of the DNA sequence. Targeted DNA methylation has been successfully documented by many groups for silencing selected genes, but recent publications have raised concerns regarding its specificity. In the current work, we developed new EpiEditors for programmable DNA methylation in cells with a high efficiency and improved specificity. First, we demonstrated that the catalytically deactivated Cas9 protein (dCas9)-SunTag scaffold, which has been used earlier for signal amplification, can be combined with the DNMT3A-DNMT3L single-chain effector domain, allowing for a strong methylation at the target genomic locus. We demonstrated that off-target activity of this system is mainly due to untargeted freely diffusing DNMT3A-DNMT3L subunits. Therefore, we generated several DNMT3A-DNMT3L variants containing mutations in the DNMT3A part, which reduced their endogenous DNA binding. We analyzed the genome-wide DNA methylation of selected variants and confirmed a striking reduction of untargeted methylation, most pronounced for the R887E mutant. For all potential applications of targeted DNA methylation, the efficiency and specificity of the treatment are the key factors. By developing highly active targeted methylation systems with strongly improved specificity, our work contributes to future applications of this approach.
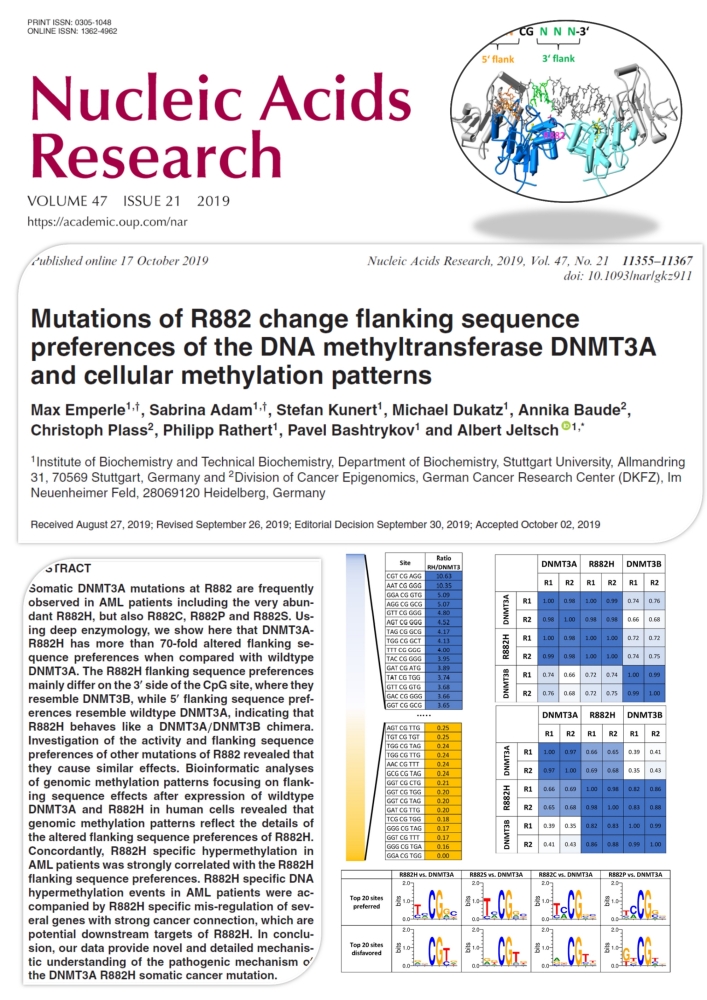
Emperle et al. (2019) Mutations of R882 change flanking sequence preferences of the DNA methyltransferase DNMT3A and cellular methylation patterns. Nucleic Acids Res. 47(21):11355-11367
Somatic DNMT3A mutations at R882 are frequently observed in AML patients including the very abundant R882H, but also R882C, R882P and R882S. Using deep enzymology, we show here that DNMT3A-R882H has more than 70-fold altered flanking sequence preferences when compared with wildtype DNMT3A. The R882H flanking sequence preferences mainly differ on the 3' side of the CpG site, where they resemble DNMT3B, while 5' flanking sequence preferences resemble wildtype DNMT3A, indicating that R882H behaves like a DNMT3A/DNMT3B chimera. Investigation of the activity and flanking sequence preferences of other mutations of R882 revealed that they cause similar effects. Bioinformatic analyses of genomic methylation patterns focusing on flanking sequence effects after expression of wildtype DNMT3A and R882H in human cells revealed that genomic methylation patterns reflect the details of the altered flanking sequence preferences of R882H. Concordantly, R882H specific hypermethylation in AML patients was strongly correlated with the R882H flanking sequence preferences. R882H specific DNA hypermethylation events in AML patients were accompanied by R882H specific mis-regulation of several genes with strong cancer connection, which are potential downstream targets of R882H. In conclusion, our data provide novel and detailed mechanistic understanding of the pathogenic mechanism of the DNMT3A R882H somatic cancer mutation.
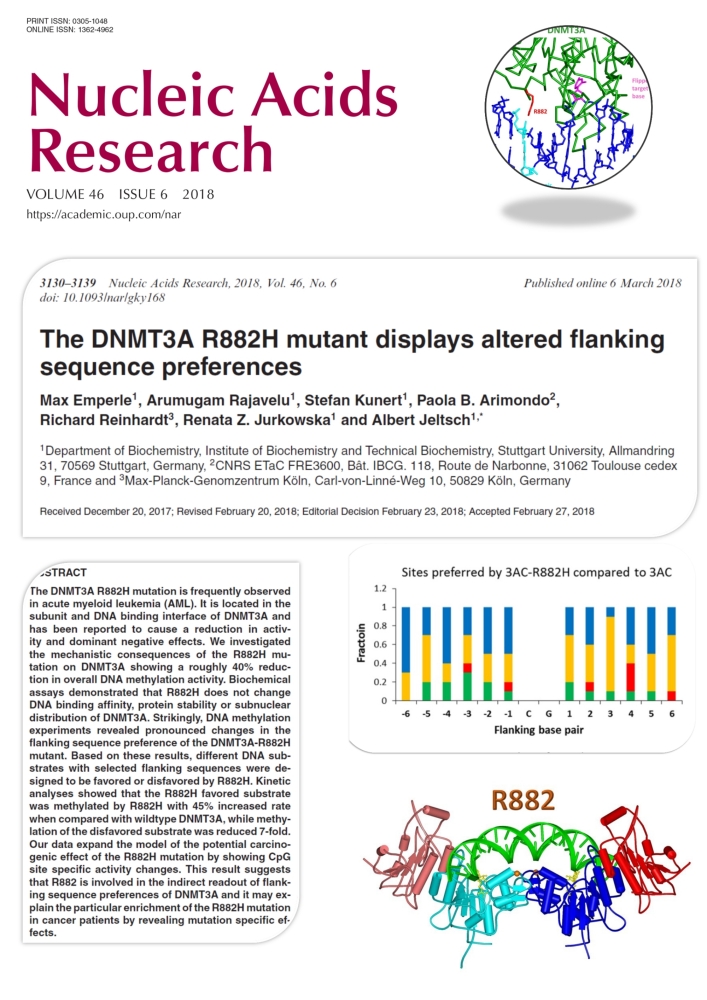
Emperle et al. (2018) The DNMT3A R882H mutant displays altered flanking sequence preferences. Nucleic Acids Res. 46(6):3130-3139
Several recent studies reported that the R882H mutation in the DNMT3A DNA methyltransferase occurs frequently in AML and it has an early role in tumorigenesis, but its exact tumor promoting mechanism is not known. DNMT3A methylates DNA at CG sites, but its activity is strongly dependent on the surrounding DNA sequence context. In this work we show that the R882H mutation causes a pronounced shift in the flanking sequence preferences of DNMT3A, indicating that some CG sites are very poorly methylated by the mutant, while others are methylated even better by the mutant than by wildtype. Our data expand the model of the potential carcinogenic effect of the R882H mutation by showing CpG site specific activity changes. This result suggests that R882 is involved in contacts of DNMT3A to the DNA backbone, which mediate an indirect readout of flanking sequence preferences. This finding may explain the particular enrichment of the R882H mutation in cancer patients.

Jurkowska et al. (2018) H3K14ac is linked to methylation of H3K9 by the triple Tudor domain of SETDB1. Nat Commun. 8(1):2057
SETDB1 is a histone methyltransferase that generates H3K9me3 marks in euchromatic regions. Here we show that the triple Tudor domain of SETDB1 binds histone H3 tails containing K14 acetylation combined with K9 methylation, and that the K9me/K14ac modification defines a novel bivalent chromatin state. Structural analyses revealed that peptide binding and K14ac recognition occurs at the interface between Tudor domains 2 and 3. Strikingly, a pocket switch mechanism was observed, because K9me1 or K9me2 is preferentially recognized by the aromatic cage of TD3, while K9me3 selectively binds to TD2. Genomic analyses show that K9me3/K14ac is enriched at SETDB1 binding sites overlapping with LINE elements, suggesting that recruitment of the SETDB1 complex to K14ac/K9me regions has a role in silencing of active genomic regions.
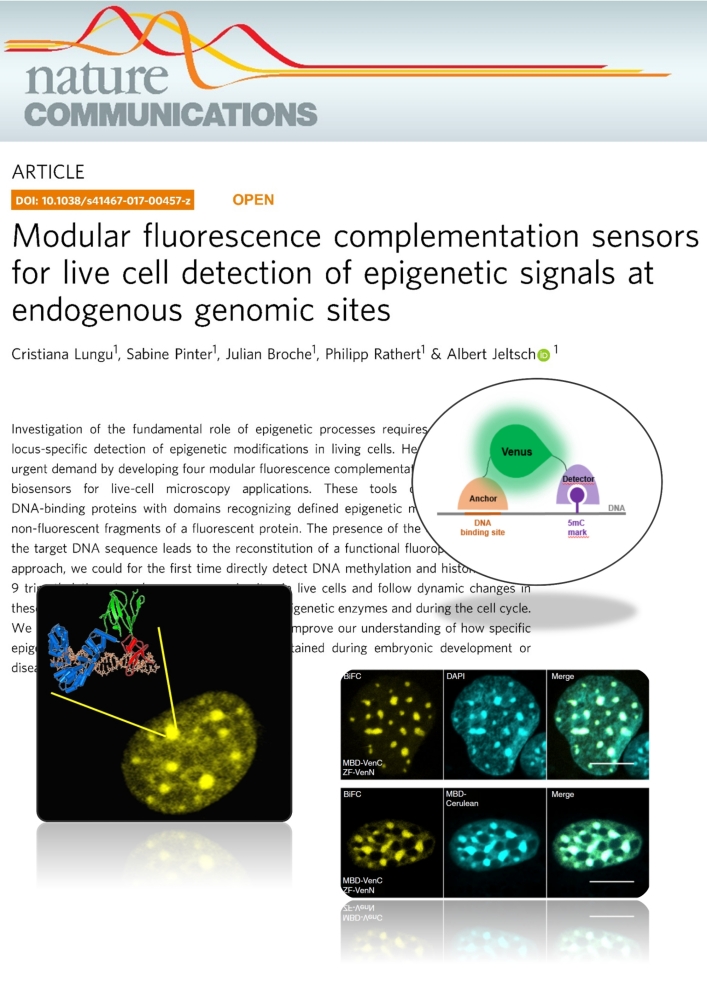
Lungu et al. (2017) Modular fluorescence complementation sensors for live cell detection of epigenetic signals at endogenous genomic sites. Nature Communications 8:649
Investigation of the fundamental role of epigenetic processes requires methods for the locus-specific detection of epigenetic modifications in living cells. Here, we address this urgent demand by developing four modular fluorescence complementation-based epigenetic biosensors for live-cell microscopy applications. These tools combine engineered DNA-binding proteins with domains recognizing defined epigenetic marks, both fused to non-fluorescent fragments of a fluorescent protein. The presence of the epigenetic mark at the target DNA sequence leads to the reconstitution of a functional fluorophore. With this approach, we could for the first time directly detect DNA methylation and histone 3 lysine 9 trimethylation at endogenous genomic sites in live cells and follow dynamic changes in these marks upon drug treatment, induction of epigenetic enzymes and during the cell cycle. We anticipate that this versatile technology will improve our understanding of how specific epigenetic signatures are set, erased and maintained during embryonic development or disease onset.Tools for imaging epigenetic modifications can shed light on the regulation of epigenetic processes. Here, the authors present a fluorescence complementation approach for detection of DNA and histone methylation at endogenous genomic sites allowing following of dynamic changes of these marks by live-cell microscopy.


Kudithipudi & Jeltsch (2016) Approaches and Guidelines for the Identification of Novel Substrates of Protein Lysine Methyltransferases. Cell Chemical Biology 23, 1049-55
Protein lysine methylation is emerging as a general post-translational modification (PTM) with essential functions regulating protein stability, activity, and protein-protein interactions. One of the outstanding challenges in this field is linking protein lysine methyltransferases (PKMTs) with specific substrates and lysine methylation events in a systematic manner. Inability to validate reported PKMT substrates delayed progress in the field and cast unnecessary doubt about protein lysine methylation as a truly general PTM. Here, we aim to provide a concise guide to help avoid some of the most common pitfalls in studies searching for new PKMT substrates and propose a set of seven basic biochemical rules: (1) include positive controls; (2) use target lysine mutations of substrate proteins as negative controls; (3) use inactive enzyme variants as negative controls; (4) report quantitative methylation data; (5) consider PKMT specificity; (6) validate methyl lysine antibodies; and (7) connect cellular and in vitro results. We explain the logic behind them and discuss how they should be implemented in the experimental work.

Jeltsch A, Jurkowska RZ (2016) Allosteric control of mammalian DNA methyltransferases - a new regulatory paradigm. Nucleic Acids Res. 44(18):8556-8575
After almost 40 years of intensive research in the DNA methylation field, we have learned a great deal about the
biochemical, structural and enzymatic properties of the mammalian DNA methyltransferases. However, the regulation
of these fascinating enzymes in cells has only begun to be uncovered. Importantly, it has been lately realized that
the precise control of DNMT activity is critically involved in the generation and maintenance of the dynamic DNA
methylation patterns in living cells. Recent crystallographic studies with DNMT1 and DNMT3A revealed that both enzymes unexpectedly undergo large domain rearrangements, which allosterically regulate their catalytic activity. This unforeseen discovery has led to the important conclusion that by influencing domain rearrangements, any PTMs or interaction partner (be it a protein, an allosteric DNA or a noncoding RNA) at various parts of the methyltransferases
could directly regulate the enzymatic activity and specificity of the DNMTs via allosteric effects, providing new and fascinating perspectives on the investigation of the effects of interactors and PTMs on these enzymes, which are presented and discussed in this review.

Kungulovski & Jeltsch (2016) Epigenome Editing: State of the Art, Concepts, and Perspectives. Trends in Genetics 32, 101-113
Epigenome editing refers to the directed alteration of chromatin marks at specific genomic loci by using targeted EpiEffectors which comprise designed DNA recognition domains (zinc finger, TAL effector, or modified CRISPR/Cas9 complex) and catalytic domains from a chromatin-modifying enzyme. Epigenome editing is a promising approach for durable gene regulation, with many applications in basic research including the investigation of the regulatory functions and logic of chromatin modifications and cellular reprogramming. From a clinical point of view, targeted regulation of disease-related genes offers novel therapeutic avenues for many diseases. We review here the progress made in this field and discuss open questions in epigenetic regulation and its stability, methods to increase the specificity of epigenome editing, and improved delivery methods for targeted EpiEffectors. Future work will reveal if the approach of epigenome editing fulfills its great promise in basic research and clinical applications.
Till 2015

Kungulovski et al. (2014) Application of histone modification-specific interaction domains as an alternative to antibodies. Genome Research 24(11): 1842-53
Post-translational modifications (PTMs) of histones constitute a major chromatin indexing mechanism, and their proper characterization is of highest biological importance. So far, PTM-specific antibodies have been the standard reagent for studying histone PTMs despite caveats such as lot-to-lot variability of specificity and binding affinity. Herein, we successfully employed naturally occurring and engineered histone modification interacting domains for detection and identification of histone PTMs and ChIP-like enrichment of different types of chromatin. Our results demonstrate that histone interacting domains are robust and highly specific reagents that can replace or complement histone modification antibodies. These domains can be produced recombinantly in Escherichia coli at low cost and constant quality. Protein design of reading domains allows for generation of novel specificities, addition of affinity tags, and preparation of PTM binding pocket variants as matching negative controls, which is not possible with antibodies.

Jeltsch & Jurkowska (2014) New concepts in DNA Methylation. Trends in Biochemical Sciences, 39(7):310-18
The widely-cited model of maintenance of DNA methylation at CpG sites implies that DNA methylation is introduced by the Dnmt3 de novo DNA methyltransferases during early development, and methylation at hemimethylated CpG sites is specifically maintained by the Dnmt1 maintenance methyltransferase. However, substantial experimental evidence from the past decade indicates that this simple model needs to be revised. DNA methylation can be described by a dynamic stochastic model, in which DNA methylation at each site is determined by the local activity of DNA methyltransferases (Dnmts), DNA demethylases, and the DNA replication rate. Through the targeting and regulation of these enzymes, DNA methylation is controlled by the network of chromatin marks.

Jurkowska & Jeltsch (2013) Genomic Imprinting—The Struggle of the Genders at the Molecular Level. Angewandte Chemie 52, 13524-36
Genomic imprinting, the parent of origin-dependent expression of genes, has been discovered as a fascinating example of the control of gene expression by epigenetic processes in the human body. It affects about 100 genes, which are often involved in growth and development. In this Review, we discuss the mechanisms leading to the generation of gender-specific imprints in form of DNA methylation marks, their preservation during growth and development of the organism, and the processes that translate parental methylation marks into monoallelic gene expression. We discuss the gender-specific dimorphic nature of imprints from an evolutionary point of view and present the prevalent model that molecular imprinting mediates a conflict of interest between the parents that occurs in viviparous animals. Finally, we summarize the relevance of parental imprinting for human health.
Jeltsch (2013) Oxygen, epigenetic signaling and the evolution of early life. Trends in Biochemical Sciences, 38(4):172-6
After approximately 3 billion years of unicellular life on Earth, multicellular animals appeared some 600 million years ago, followed by the rapid emergence of most animal phyla during the Cambrian radiation. This evolutionary jump was paralleled by an increase in atmospheric oxygen, which I propose allowed the generation of epigenetic signaling systems that are essential for cellular differentiation in animals. Epigenetic signaling is based on the reversible deposition of chemically stable marks in DNA and histone proteins, with methylation of cytosine and lysine residues, respectively, playing a central role. Recent evidence indicates that the removal of such methyl groups critically depends on oxygenases. Hence, reversible epigenetic systems could only appear after accumulation of oxygen in the atmosphere.

Dhayalan et al. (2011) Specificity analysis based identification of new methylation targets of the SET7/9 protein lysine methyltransferase. Chemistry & Biology 18, 111-120
We applied peptide array methylation to determine an optimized target sequence for the SET7/9 (KMT7) protein lysine methyltransferase. Based on this, we identified 91 new peptide substrates from human proteins, many of them better than known substrates. We confirmed methylation of corresponding protein domains in vitro and in vivo with a high success rate for strongly methylated peptides and showed methylation of nine nonhistone proteins (AKA6, CENPC1, MeCP2, MINT, PPARBP, ZDH8, Cullin1, IRF1, and [weakly] TTK) and of H2A and H2B, which more than doubles the number of known SET7/9 targets. SET7/9 is inhibited by phosphorylation of histone and nonhistone substrate proteins. One lysine in the MINT protein is dimethylated in vitro and in vivo demonstrating that the product pattern created by SET7/9 depends on the amino acid sequence context of the target site.

Bock et al. (2011) Detailed specificity analysis of antibodies binding to modified histone tails with peptide arrays. Epigenetics 6, 265-263
Chromatin structure is greatly influenced by histone tail post-translational modifications (PTM), which also play a central role in epigenetic processes. Antibodies against modified histone tails are central research reagents in chromatin biology and molecular epigenetics. We applied Celluspots peptide arrays for the specificity analysis of 36 commercial antibodies from different suppliers which are directed towards modified histone tails. The arrays contained 384 peptides from 8 different regions of the N-terminal tails of histones, viz. H3 1-19, 7-26, 16-35 and 26-45, H4 1-19 and 11-30, H2A 1-19 and H2B 1-19, featuring 59 post-translational modifications in many different combinations. Using various controls we document the reliability of the method. Our analysis revealed previously undocumented details in the specificity profile. Most of the antibodies bound well to the PTM they have been raised for, but some failed. In addition some antibodies showed high cross-reactivity and most antibodies were inhibited by specific additional PTMs close to the primary one. Furthermore, specificity profiles for antibodies directed towards the same modification sometimes were very different. The specificity of antibodies used in epigenetic research is an important issue. We provide a catalog of antibody specificity profiles for 36 widely used commercial histone tail PTM antibodies. Better knowledge about the specificity profiles of antibodies will enable researchers to implement necessary control experiments in biological studies and allow more reliable interpretation of biological experiments using these antibodies.

Jeltsch (2010) Phylogeny of Methylomes, Science 328, 837-8
The DNA of most species is methylated, containing the modified base 5-methylcytosine. This modification has a role in silencing gene expression, among other important functions. Advances in sequencing methods have allowed measurement of the first complete genome-wide DNA methylation map (“methylome”) of the model plant Arabidopsis thaliana and human cells. Studies by Feng et al. and by Zemach et al. on page 916 of this issue now expand this list by providing genome-wide methylomes for 20 additional species, revealing important conserved features and phylogenetic relationships of the methylation machinery.

Rathert et al. (2008) Protein lysine methyltransferase G9a acts on non-histone targets. Nat. Chem. Biol. 4, 344-6
By methylation of peptide arrays, we determined the specificity profile of the protein methyltransferase G9a. We show that it mostly recognizes an Arg-Lys sequence and that its activity is inhibited by methylation of the arginine residue. Using the specificity profile, we identified new non-histone protein targets of G9a, including CDYL1, WIZ, ACINUS and G9a (automethylation), as well as peptides derived from CSB. We demonstrate potential downstream signaling pathways for methylation of non-histone proteins.

Rathert et al. (2008) Analysis of the Substrate Specificity of the Dim-5 Histone Lysine Methyltransferases Using Peptide Arrays. Chemistry & Biology 15, 5-11
Histone methylation is an epigenetic mark essential for gene regulation and development. We introduce peptide SPOT synthesis to study sequence specificity of the Dim-5 histone-3 lysine-9 methyltransferase. Dim-5 recognizes R8-G12 of the H3 tail with T11 and G12 being the most important specificity determinants. Exchange of H3 tail residue S10 and T11 by E strongly reduced methylation by Dim-5, suggesting that phosphorylation of S10 or T11 may regulate the activity of Dim-5. In the Dim-5/peptide structure, E227 interacts with H3R8 and D209 with H3-S10. Mutations of E227 or D209 caused predictable changes in the substrate preference, illustrating that peptide recognition of histone methyltransferases can be altered by protein design. Comparative analyses of peptide arrays with wild-type and mutant enzymes, therefore, are well suited to investigate the target specificity of protein methyltransferases and study epigenetic crosstalk.

Jia et al. (2007) Structure of the Dnmt3L-Dnmt3a Complex Suggests a Model for de novo DNA Methylation. Nature 449, 248-51
Genetic imprinting, found in flowering plants and placental mammals, uses DNA methylation to yield gene expression that is dependent on the parent of origin. DNA methyltransferase 3a (Dnmt3a) and its regulatory factor, DNA methyltransferase 3-like protein (Dnmt3L), are both required for the de novo DNA methylation of imprinted genes in mammalian germ cells. Dnmt3L interacts specifically with unmethylated lysine 4 of histone H3 through its amino-terminal PHD (plant homeodomain)-like domain. Here we show, with the use of crystallography, that the carboxy-terminal domain of human Dnmt3L interacts with the catalytic domain of Dnmt3a, demonstrating that Dnmt3L has dual functions of binding the unmethylated histone tail and activating DNA methyltransferase. The complexed C-terminal domains of Dnmt3a and Dnmt3L showed further dimerization through Dnmt3a-Dnmt3a interaction, forming a tetrameric complex with two active sites. Substitution of key non-catalytic residues at the Dnmt3a-Dnmt3L interface or the Dnmt3a-Dnmt3a interface eliminated enzymatic activity. Molecular modelling of a DNA-Dnmt3a dimer indicated that the two active sites are separated by about one DNA helical turn. The C-terminal domain of Dnmt3a oligomerizes on DNA to form a nucleoprotein filament. A periodicity in the activity of Dnmt3a on long DNA revealed a correlation of methylated CpG sites at distances of eight to ten base pairs, indicating that oligomerization leads Dnmt3a to methylate DNA in a periodic pattern. A similar periodicity is observed for the frequency of CpG sites in the differentially methylated regions of 12 maternally imprinted mouse genes. These results suggest a basis for the recognition and methylation of differentially methylated regions in imprinted genes, involving the detection of both nucleosome modification and CpG spacing.

Horton et al. (2005) Transition from non-specific to specific DNA interaction along the DNA recognition pathway of DAM methyltransferase. Cell 121, 349-61
DNA methyltransferases methylate target bases within specific nucleotide sequences. Three structures are described for bacteriophage T4 DNA-adenine methyltransferase (T4Dam) in ternary complexes with partially and fully specific DNA and a methyl-donor analog. We also report the effects of substitutions in the related Escherichia coli DNA methyltransferase (EcoDam), altering residues corresponding to those involved in specific interaction with the canonical GATC target sequence in T4Dam. We have identified two types of protein-DNA interactions: discriminatory contacts, which stabilize the transition state and accelerate methylation of the cognate site, and antidiscriminatory contacts, which do not significantly affect methylation of the cognate site but disfavor activity at noncognate sites. These structures illustrate the transition in enzyme-DNA interaction from nonspecific to specific interaction, suggesting that there is a temporal order for formation of specific contacts.
Contact

Albert Jeltsch
Prof. Dr.Head of Biochemistry Department and Acting Director of the IBTB